ProstOmics - Department of Circulation and Medical Imaging
ProstOmics

Read more about our methods
Tissue biobanking
Tissue biobanking
We analyse a large set of fresh frozen prostate cancer and whole-mount tissue obtained from Biobank1. We have also collected over 2000 prostatectomy needle biopsies samples from 1000 patients. All samples are frozen and stored in liquid nitrogen or at -80 °C to minimize tissue degradation. Samples are handled in a clean and RNAse-free environment to prevent contamination and ensure conservation to allow optimal high-quality omics-data collection.
We have developed a standardized and targeted tissue sampling technology prototype, called «Augmented Reality enhanced and automated Tissue Sampler (ARTS). ARTS allowed us to collect over thousand sub-samples from the fresh frozen whole-mount tissue slices of over hundred prostate cancer patients that are specifically tailored to our research interest.


Histopathology
Histopathology
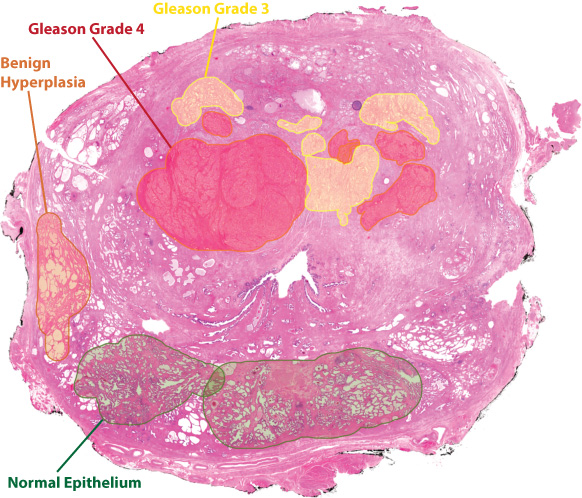
Histopathology involves microscopically evaluating stained tissue material to identify abnormalities indicative of cancer. This is an invaluable part of the standard clinical evaluation and plays a key role in our work.
First, we use histopathology from adjacent formalin fixed tissue to identify areas of interest (see 'Tissue biobanking') to collect sub samples (see illustration below). These sub samples are then sectioned and stained, and a new round of histopathology is performed to annotate different tissue regions (for example cancer, no cancer) to allow classification of data obtained with imaging methods such as mass spectroscopy imaging (MSI).
We are collaborating with experienced uropathologists, Elin Richardsen (UiT, Tromsø Universitetssykehus), Øystein Størkersen and Trond Viset (St. Olavs Hospital), who perform all histopathology evaluations.

DNA Methylation
DNA Methylation
DNA methylation is a mechanism in which genes are switched on and off by the addition of methyl groups (CH3) to cytosines. This can both encourage and discourage transcription thus directly affect whether a gene gets transcribed or not. In addition, DNA methylation can affect chromatin structure, to either pack the DNA very tightly for storage, or to unwind and open the DNA for easier access for the transcriptional machinery. DNA methylation is one of the earliest molecular aberrations observed in cancer development, and our hypothesis is that DNA methylation markers in prostate tissue can reveal cancer development and risk of recurrence at an early stage.
We measure DNA methylation by use of an array technology, more specifically the Illumina EPIC platform, at the Genomics Core Facility (GCF, NTNU).
Microproteomics
Microproteomics
Laser microdissection is a method for isolating cell populations and tissue areas of interest. The dissected samples can further be analysed by various analytical methods, such as genomics, transcriptomics and proteomics workflows. We have analysed prostate tissue micro-dissected samples by an optimized LC-MS/MS protocol for proteomics. From these dissected samples, we have identified 2500 proteins from 2000 cells and 600 proteins from 250 cells. This microproteomic workflow will be used to determine the proteomic profiles in different tissue types, such as normal and cancerous tissue. Our goal is to identify proteins that potentially can be used as diagnostic and prognostic biomarkers for prostate cancer.
These analyses are done in collaboration with the Proteomics and Modomics Experimental Core Facility (PROMEC, NTNU).

Transcriptomics
Transcriptomics
Transcriptome profiling is the study of the complete set of RNA transcribed by a given cell population. RNA sequencing is the current standard approach for transcriptome profiling. It offers the opportunity to study many aspects of the transcriptome such as gene fusions, isoforms and non-coding RNA. RNA-seq provides a robust snapshot of the cellular state of a sample through its RNA-profile. In addition, the transcriptome profile of samples can be compared against the huge number of publicly available transcriptome signatures and gene-sets using, for example, Gene Set Enrichment Analysis (GSEA). Transcriptome profiling thus provides a functional hub for the integration of data from other protocols (MALDI imaging, HR MAS MRS, mpMRI) with the existing knowledge on prostate cancer from the literature.
We use both bulk and spatial RNA-seq for transcriptome profiling. The bulk RNA-seq approach is based on the average gene expression from a cell population, whereas the spatial RNA-seq technology maps where the gene expression in a tissue sample is occurring. To study spatial gene expression, we use the Visium Spatial Gene Expression assay from 10x Genomics. The spatial transcriptome data is layered onto histologically stained images and other omics-data to examine cells in their microenvironment (see figure).
RNA isolation and sequencing is done in collaboration with biobank1 (St. Olavs) and the Genomics Core Facility (GCF) at NTNU.
Metallomics
Metallomics
The healthy prostate, or more precisely the peripheral zone of the prostate has a much higher zinc concentration than other soft tissue in the human body. This is also reflected in the prostatic fluid, which contains about 500-times more zinc than the blood plasma.
Interestingly, zinc concentration - together with citrate - is clearly reduced in prostate cancer. We aim to better understand the role of zinc, its transporters and potentially other metals/cations in prostate cancer. We hypothesize that zinc and/or its transporters might serve as biomarkers for prostate cancer aggressiveness and/or recurrence. Thus, we investigate the distribution and concentration of zinc, zinc-related proteins and other metals/cations in a big cohort of human prostate tissue by using modern fluorescence microscopy, laser ablation inductively coupled plasma (LA-ICP) and matrix-assisted laser desorption/ionization (MALDI) mass spectroscopy imaging (MSI). We hope that identified patterns might help to better diagnose the individual prostate cancer risk from standard needle biopsies or even from so called liquid biopsies (prostate secrete, urine, blood serum samples).
MALDI-imaging
MALDI-imaging
With mass spectrometry imaging (MSI) several spectra are acquired in a pixel wise matter from a tissue section. This makes it possible to analyze measurements that are specific for different tissue types. MALDI MSI is the most common method for this purpose. MSI is of great value within our research focus as prostate cancer is a heterogeneous disease with different tissue types, such as normal epithelium, stroma and various grades of cancer epithelium, occurring in the same tissue sample.
We are using MALDI MSI for analysis of small metabolites, lipids and trypsinated peptides in collaboration with the M4I group at Maastricht University and MR Core Facility (NTNU). We use a RapifleX MALDI Tissuetyper (MALDI-TOF/TOF) located MR Core Facility and multiple MS imaging machines at Maastricht University.

Contact
![]()
![]()
Project user representative
 |
Ola Kindseth, sociologist (retired)Relevant experience: |